


2020-08-24 by Quick Biology Inc.
Ticks (Acari: Ixodidae) are obligate blood-feeding arthropods. They are the most versatile vectors, transmit the most diverse human and animal pathogens. Due to urbanization, deforestation, and climate change, tick-borne diseases (TBDs) have increased unpredictably, worsen the quality of human health. In recent Cell, researchers in the Chinese Academic of Sciences published their work of six high-quality ixodid tick genomes and 678 re-sequenced tick specimens online (ref1. Fig.1).
To generate six high-quality Ixodid Tick reference genomes, they first constructed DNA libraries of 15 kb or more for the Long-Read sequencing (PacBio Sequel System) and generated 162-303 Gb of subreads with high sequencing depth (approximately 67-95X); To do error-correction of PacBio reads, they further constructed short-fragment libraries (350 bp) for WGS and sequenced them using the Illumina HiSeq X-Ten platform. These high-quality short reads were used to perform K-mer frequency analyses to estimate the genome sizes and to correct the short insertions or deletions (indels) and substitutions in the PacBio assembly. To further improve the continuity of the assembled tick genomes and anchor the assemblies into chromosomes, Hi-C data was used to order and orient the contigs as well as to correct mis-joined sections and merge overlaps (Fig. 2). Finally, they assembled six tick genomes, achieving 8,620–15,174 contigs with scaffold N50 lengths of 533-208,696 kb and contig N50 lengths of 340–1,800 kb. By combining transcriptome-Based (RNA-seq data), homology-based, and ab initio approaches, 25,718–29,857 protein-coding genes were predicted from these tick genomes.
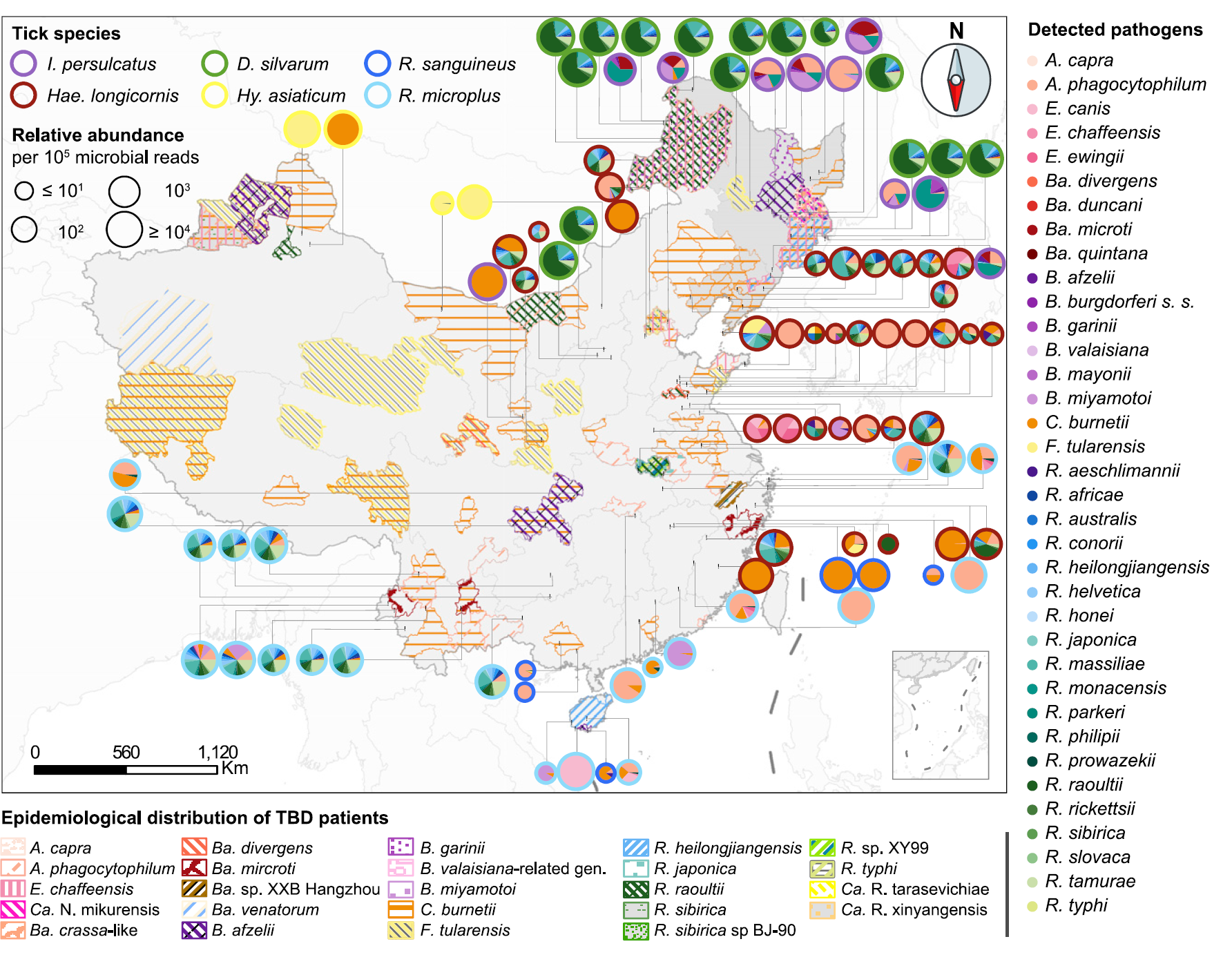
This large cohort resequencing is a great resource to understand the genetic diversity, population structure (Fig.2). Particularly, researchers by metagenome analyses investigated pathogen composition and distributions in tick specimens (Fig.3). They also identified species-specific determinants associated with different host ranges and life cycles, which provides us an invaluable resource for research and control of ticks and tick-borne diseases.
Fig. 1: Comprehensive genome analysis and pathogen composition of Six Tick species. (Cover image from ref1)
Fig. 2: Genomic Comparison of Six Tick Genomes. (A) Map of sample collection. (B) Tick’s 3-host life cycles. (C) Hi-C interactive heatmap of chromosomes for five ticks. (D) Phylogenetic tree of six tick species. (from ref1)
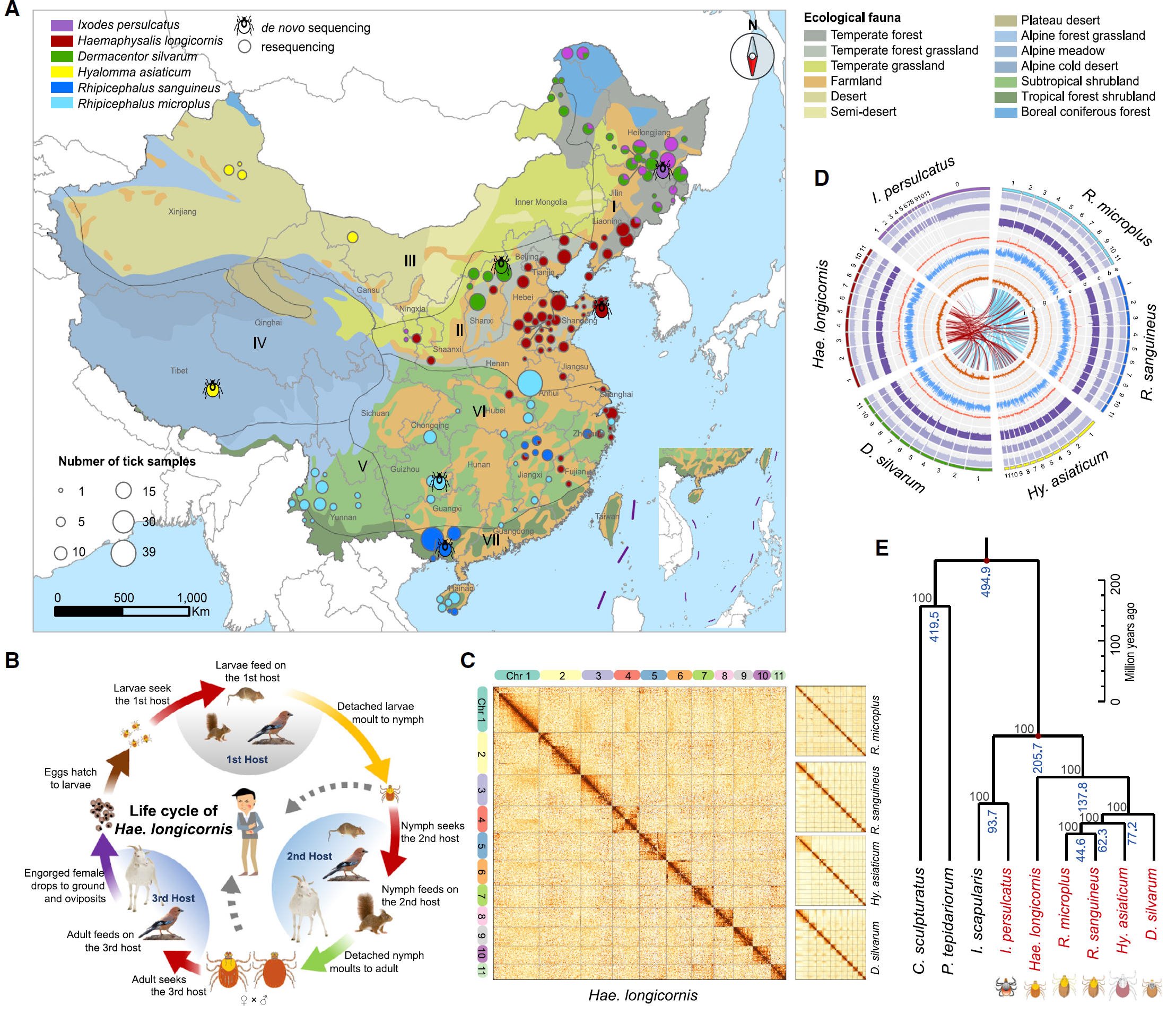
Fig. 3: Potential Pathogen Profiling of Six Tick Species. Epidemiological distribution of tick-borne disease (TBD) patients and tick pathogens. The cases of human infection were reported between 1980 and 2020. The pies indicate pathogen composition, with the color of circle outlines representing tick species. The circle size indicates the relative abundance of all pathogens per 105 microbial reads, and the color and area of the pies indicate the species and relative abundance of each pathogen, respectively. Northeastern China is highlighted in dark gray. (from ref1)
Quick Biology is an expert in Whole-genome sequencing and de novo genome assembly, we aim to unleash the power of NGS in human diseases, make pathogen genomics into practice. Find More at Quick Biology.
Ref:
1. Large-Scale Comparative Analyses of Tick Genomes Elucidate Their Genetic Diversity and Vector Capacities. 2020 Cell 182 1-13. https://www-sciencedirect-com.ezp-prod1.hul.harvard.edu/science/article/pii/S0092867420309314