


02-22-2021 By Quick Biology
Genome-wide association studies (GWAS) have implicated hundreds of thousands of single-nucleotide polymorphisms (SNPs) in human diseases and traits, but very few of them have been functionally characterized. Most of these disease or trait-associated SNPs are in the intron region (i.e., non-coding region) or intergenic region, which makes it harder to interpret whether they are true causal genetic sites or just hitch-hiking genetic variants.
In Nature, Dr. Bing Ren Lab, and Dr. Jussi Taipale Lab from the University of California San Diego, focusing on gene regulation at the epigenetic level, systematically assess the binding of 270 human transcription factors to 95,886 noncoding variants in the human genome. By using an ultra-high-throughput multiplex protein-DNA binding assay, termed single-nucleotide polymorphism evaluation by systematic evolution of ligands by exponential enrichment (SNP-SELEX), they examine in vitro binding of human transcription factors to common sequence variants using a sampling scheme that surveys candidates cis-regulatory variants near the reported type-2 diabetes (T2D) risk loci (Fig.1, ref1). Not like traditional SELEX using randomized DNA sequences as input, in SNP-SELEX, they used a library of 40-bp DNA matching the reference human genomic sequence, with the center position corresponding to tested SNPs permutated to all four bases (Fig.2). By systematic analysis of SNP-SELEX, they estimate the relative affinity of these transcription factors to each variant in vitro. They report highly predictive models for 94 human transcription factors and demonstrate their utility in genome-wide association studies and understanding of the molecular pathways involved in diverse human traits and diseases. See more in their GVAT (Genetic Variants Allelic TF Binding Database) website (http://renlab.sdsc.edu/GVATdb/).
Figure 1: High-throughput analysis of the binding of human transcription factors to common sequence variants by SNP-SELEX. a, An overview of the SNP-SELEX experimental procedure. N indicates the position of SNPs. FL, full length; QC, quality control; TFs, transcription factors. b, The data obtained from each SELEX cycle were analyzed to determine OBS and PBS. Two alleles of the SNP are shown, the reference allele (triangle) and the alternative allele (circle). Differential binding information for all SNPs tested is publicly available from the GVAT database. c, d, Histograms show the number of pbSNPs bound by each transcription factor (c), and the number of transcription factors showing allelic binding for each pbSNP (d). e, A clustering diagram of transcription factors tested in this study was generated on the basis of the pairwise Pearson correlation of their DNA binding specificity from the SNP SELEX data. For each pair of experiments, we computed the Pearson correlation coefficient (PCC) and dissimilarity (1 − PCC) of PBS between significantly enriched oligonucleotides in both experiments and clustered them using the UPGMA algorithm.
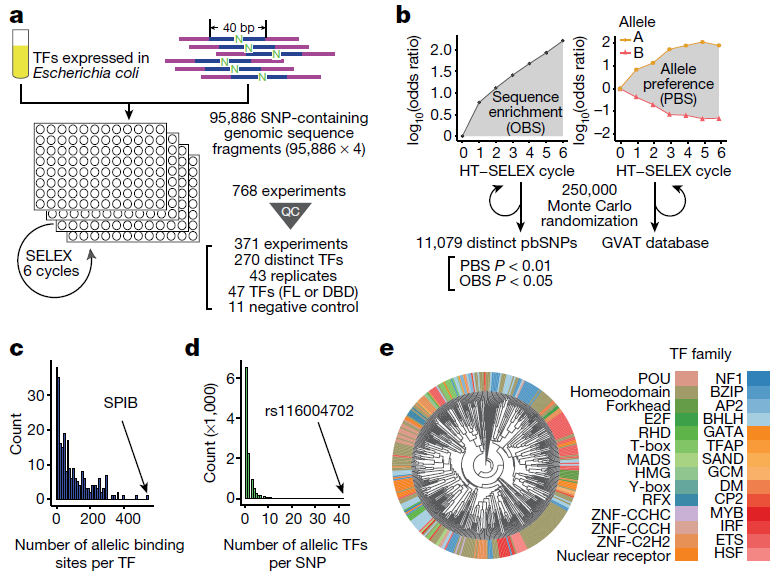
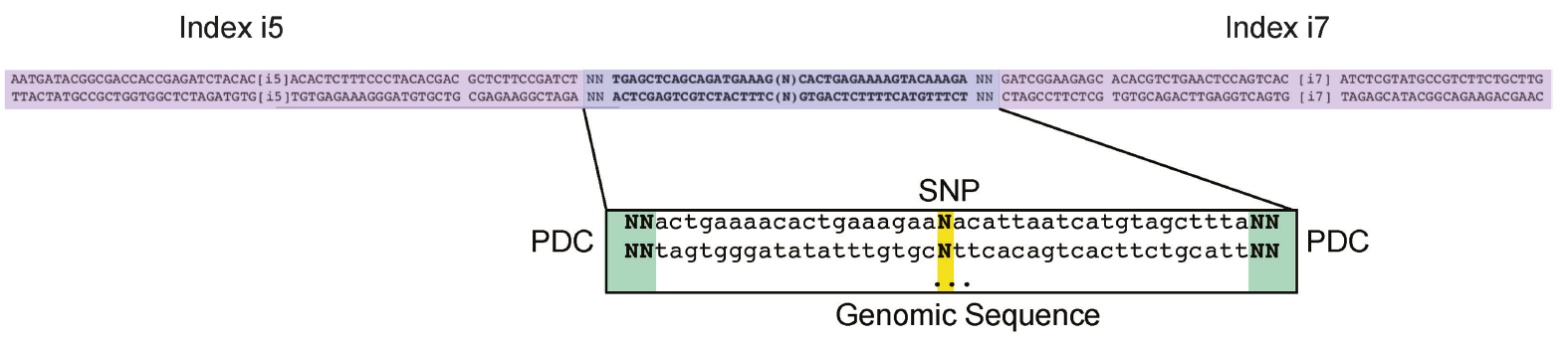
Figure 2: The sequence features of input oligonucleotides. Two random nucleotides were added to each end of the oligos as unique molecule identifiers (UMIs) to remove over-represented PCR duplicates. Illumina TruSeq dual-index system was adapted for oligo design.
Quick Biology can assist you with NGS screening design and NGS sequencing. Find More at Quick Biology.
See resource:
1. Yan, J. et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature (2021). doi:10.1038/s41586-021-03211-0